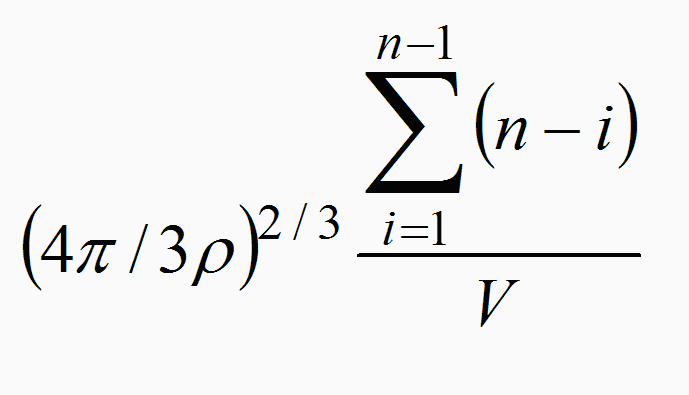©2014
Federico I. Talens-Alesson
FOREWORD
What we are going to discuss here is whether a charged surface
changes the solubility of a salt. If you have followed my blogs, you
know that I postulate that solution chemistry is (mostly) the
chemistry between of surface excesses of chemicals. A normal boundary
to a solution is either the air/water surface or a wall (the
container's) or a probe. However, seed crystals and colloidal
particles would also be surfaces against which a surface excess may
develop. This is the underlying reason for all sorts of fouling and
cryytal growth/nucleation phenomena.
SURFACE EXCESSES IN SURFACTANT
SYSTEMS
In the particular case of a micellar solution of an ionic surfactant
(which is a type of colloidal system), it also demands saturation of
the air/water interface with surfactant (which means that it also
demands saturation of any other surface) forming a charged surface
layer. At the time I was still publishing in scientific journals, and
I went as far as to show that the critical conditions for
micellisation is ionic pairing between the surface excess of an
ionic surfactant and its counter ion's surface excess
(Talens-Alesson, Langmuir 2010, 26(22), 16812–16817)
However, as the surface excess and concentration are related, I also
showed that using Bjerrum's correlation it is possible to estimate
the relative bindings of different polyvalent cations onto anionic
surfactant micelles. Biding of cations onto anionic micelles is a
particular case of ionic pairing, and ion pairing is key to
precipitation and other reactions between electrolytes. It is through
forming of ionic pairs that the building blocks for crystal assembly
take place (Talens-Alesson, J. Phys. Chem. B 2009, 113, 9779–9785).
In short, the higher charge counter ion predominates and for as long
as the half length of the hedge of a cube of volume (volume of
solution/concentration of higher charge counter ion times Avogadro's
Number) is less than Bjerrum's distance for ionic pairing, then this
counter ion would bind onto the micelle. This works well for the
binding of Al3+ and Zn2+ and subsequent
flocculation of the surfactant SDS (sodium dodecylsulfate).
This required assigning an apparent charge of 5e- to the
“counter ion adsorption patch” of an SDS micelle. This was
required even though the binding of counter ions onto an SDS
spherical micelle follows a ratio 1 counter ion (regardless of
charge) per three SDS molecules in the micelle. This means that the
binding attraction is more than it should be expected. And this
brings us to the question of whether enhanced interaction of
electrolytes near micelles is possible.
In
practice, research on ionic surfactants focuses on how they help
other chemicals (essentially organic ones) to dissolve through
micellization. It also focuses on the solubility of ionic surfactants
themselves in the presence of a range of counterions. The latter has
to do with the presence of certain cations available (and therefore
in soluble form) either in nature (soils, for remediation or oil
recovery, for example) or in water in technological applications
(detergency). Of course, how the surfactant may affect the solubility
of something already precipitated seems a rather bizarre subject of
research, particularly if one believes that chemical reactions in
solution are bulk phase phenomena. Of course, I don't. And it should
not be so difficult to realize that a broader investigation of how
micelles affect chemical reactions should take place, considering
that there is some evidence in its favor. An example of a
micelle-enhanced Fenton reaction is given by Talens-Alesson (Chem,
Eng Technol 2003, 26(6) 684-687).
CASE 1. EARLY EVIDENCE FOR HIGHER THAN EXPECTED COUNTER ION BINDING
ONTO IONIC MICELLES.
Let us consider an aged data set, dating back to 1994, by a team from
the IAST at the University of Oklahoma in Norman (Scamehorn et al.,
Separation Sci Technology, 1994, 29(7) 809-830). It describes binding
of single cations and their mixtures onto micelles of anionic
surfactant SDS.

In Figure 1 I plot my own calculation for experimental binding ratios
for the cations investigated by the researchers at IAST, following:
total cation concentration minus final cation concentration in
solution
over
one third the difference between total SDS concentration minus final
SDS concentration in solution
together with my own predicted 'Bjerrum's” binding ratio as
discussed above. There is a constant offset between the actual
binding and the predicted one using Bjerrum's correlation.
If Bjerrum's correlation was correct, then binding would be lower
than it actually is. But then, Bjerrum's correlation is derived on
the assumption that solution chemistry is bulk phase concentration
chemistry. Therefore, it is possible that there is a condition for
diluted concentrations still matching the true critical ionic pairing
condition at the surface excess regions. But this means that ionic
pairing can occur well below Bjerrum's condition for ionic pairing:
it would occur for the “surface excess” condition for ionic
pairing, which would be masked by Bjerrum's use of bulk phase
concentration.
This deviation is consistent with the fact that above saturation the
surface excess is constant: above the saturation surface excess the
value is independent of the concentration, and therefore the
condition for ionic pairing appears to be a constant. The additional
binding at a lower concentration would be a constant amount, related
to the difference between the saturation surface excess and the
critical surface excess for ionic pairing. Figure 2 shows this.

This does not imply that the linear relationship can be stretched
indefinitely: Figure 3 shows some data by IAST researchers for 4mM
counter ion concentrations, below a Bjerrum's concentration (would be
4.5 mM for a divalent cation against an SDS micelle). In these
experiments the variation is in the SDS concentration: from 100 mM,
to 200 mM and on to 400 mM. The concentration of counter ion is
always 4 mM. As the concentration of surfactant increases, the
binding drops which is consistent with the fact that there is more
surfactant colloidal surface for the same amount of counter ion. As
the ratio surface to volume increases, with the ratio surface excess
to concentration being basically constant at low concentrations, then
the total amount of solute is distributed over a lower surface excess
matching a lower concentration. The binding ratio decreases but the
total binding slightly increases. Binding of counterions onto ionic
micelles may therefore exceed theoretical values and micelles are
attractors of counter ions.

CASE 2. IONIC PAIR-DRIVEN INCORPORATION OF ELECTROLYTES ONTO AL3+/SDS
MICELLAR FLOCCULATES.
Paton and Talens (Langmuir, 2002, 18 (22), pp 8295–8301) describe
competitive binding of Al3+ and Zn2+ onto flocculating SDS micelles.
The results indicate that the total charge of cations exceeds that of
surfactant in the floc in some cases, with apparent charge ratios
higher than 1 and sometimes up to 1.8 (Figure 4). The only
explanation is ionic pairing between said cations and sulfate anions
present in the solution. This leaves them as lower charge species and
more of them are required to reach electroneutrality around the
micelles.

This means that the flocculate is a mixture AlxZnyDSzSO4t. It is
important to notice that the concentrations of all the chemicals are
below their solubility limits. Aluminum sulfate and zinc sulfate are
soluble at higher concentrations than the ones the flocculate forms.
The excess chemicals are “opted in” into the flocs irrespective
of their solubilites.
This means that a discontinuity in the solution, with the ability to
cause enhanced adsorption of counter ions, may lead to enhanced
capture of ionic pairs/salts. In a subsequent paper it was proposed
that ionic pairs AlSO4+ and ZnSO40
may co-adsorb onto the micelles (Talens-Alesson, J. Phys. Chem. B
2009, 113, 9779–9785), explaining this apparent charge inversion.
However, this explanation does not change the fact that well below
their saturation conditions, fragments belonging to the Al2(SO4)3
and ZnSO4 salts are incorporated onto the flocs.
CASE 3. QUESTION MARK EXPERIMENT.
This
is a peculiar experimental result, as it is reported as part of a
fraudulent effort consisting in two papers in which staff at the
University of Nottingham (one of them eventually moving to Oxford)
plagiarized
work of mine to pretend that they were active in the research of
micellar flocculation.
In the second of them, published in Separation
Purification Technol (2008) they claim flocculation of AL(DS)3
from solutions in which the residual Al and SDS concentrations they
claim to have found (far below normal results in micellar
flocculation of Al(DS)3) could only be explained by a
strange charge ratio of around 4/1 between Al and DS (assuming the
concentrations given where Al3+ and not aluminum sulfate,
in which case it would be around 8/1). While in pH adjusted solutions
it is possible to have rather high ratios Al3+/SDS (2 to
1), this only happens because in the adequate range of pH Al13
is present. The authors of this document do not state such pH
adjustement, and the concentrations of surfactnat and SDS do not
justify the charge inversion, as observed in previous work. A number
of documents on the case of this story on plagiarism and fraud are
available on the net.
However, there is a question: considering that various other
statements make absolute no sense (stating that benzoic acid is a
reagent for the analysis of anionic surfactants in two phase
titration, that the formula of aluminum sulfate is AlSO4,
or that phenol is an alkali, or that the Critical Micelle
Concentration of a surfactant can ever be tolerable in a water
targeted for processing for drinking purposes) even though the result
seems unlikely there is no obvious reason why the authors, in their
overall ignorance, should realize that the expected Al concentration
would be unreasonably high considering they expect to remove a
concentration of phenol which would be unrealistic (too low for an
attempt to recycle/recovery). In principle this should be an
irrelevant question if the experiment was a regular micellar
flocculation one.
But there is a peculiarity in this experiment. Unlike previous work,
it was carried out while air was bubbled through the solution as
flocculation took place. This presents an interesting question. It is
known that foam can be used to remove the foaming surfactant but also
other chemicals present, adsorbed on the surfactant layer of the
foam. There is for example work on the removal of moderate amounts of
phenol in SDS foams.
What if the foam actually provides a barrier that may be crossed by
ionic pairs or ionic molecules? What if on the air side they can
remain for long enough for a shift in the effective equilibrium of
solution, becoming an electrolyte sink? Figure 5 illustrates the
idea.

CONCLUSION.
On the grounds of previous information it is suggested that foams may
assist to cause an “enhanced” insolubilistion of salts, which a
variety of potential technological applications.
The way in which this may be brought to happen would be to use a micellar solution of surfactant, and cause air to bubble in the presence of the electrolytes targeted to produce the desired insoluble salt.